一种超细氧化镧的可控制备方法与流程
1.本发明涉及氧化镧制备技术领域,特别是涉及一种超细氧化镧的可控制备方法。
背景技术:
2.氧化镧是轻稀土中的重要产品之一。微溶于水,易与酸反应,生成相应的盐。易与空气中的二氧化碳和水发生反应,逐渐变成碳酸镧。如今,具有良好物理化学性能的氧化镧产品,在军工、民用和高科技领域都获得了广泛的应用。如,镧光学玻璃是以la2o3为主要成分的硼酸盐或硼硅酸盐的高级光学玻璃,具有高折射和低色散的独特光学特性;在电子陶瓷中加入la2o3后,可改善陶瓷的烧结性、致密性、纤维结构和相组成等,能够明显提高陶瓷的应用性能;la2o3还可以用作汽车尾气净化。
3.发明专利cn 105084407 a公开了一种超细氧化镧及其制备方法,制备过程中使用了具有毒性的草酸作为沉淀剂,其制备的氧化镧粒径d50为1.2-1.5μm,其制备的粒径比较大。
4.发明专利cn 105948098 a公开了一种球形氧化镧,采用pvp作为表面活性剂,短链有机酸影响球形颗粒的尺寸,制备的球形氧化镧的平均粒径为150nm-1.2μm,反应时需要控制在160-240℃下,反应温度较高。
5.发明专利cn 110790297 a公开了一种氧化镧的生产工艺,该发明以乙二醇和聚乙二醇600为溶剂,以聚乙烯吡咯烷酮为表面活性剂,制备的氧化镧粒径0.05-10μm范围内,其粒径最小的控制在0.05微米,无法制备更小尺寸的氧化镧。
技术实现要素:
6.针对上述现有技术中存在的问题,本发明的目的是提供一种超细氧化镧的可控制备方法,可以根据不同的需要制备不同粒径的氧化镧。
7.为实现上述目的,本发明通过以下技术方案来实现:一种超细氧化镧的可控制备方法,包括以下步骤:s1:将氧化镧加入到酸性溶液中制备镧盐溶液;s2:将镧盐溶液加入到乙二醇、聚乙二醇800的混合溶液中,搅拌混合后加入ctab和nacl溶液;s3:将步骤s2的混合液加热至60-80℃,并缓慢加入nh4hco3溶液,搅拌混均,然后加入氨水调节ph=5-7,搅拌1-3h,使溶液中出现白色沉淀物,过滤、洗涤、干燥得到粉末状碱式碳酸镧laohco3;s4:将碱式碳酸镧在800-900℃的马弗炉中煅烧2-6h,即得超细la2o3粉末。
8.进一步地,步骤s1中所述酸性溶液为硝酸,浓度为0.5-2.0mol/l;所述镧盐为硝酸镧,浓度为0.5-1.5mol/l。
9.进一步地,步骤s2中乙二醇、聚乙二醇800的混合溶液中乙二醇与聚乙二醇800的混合体积比为1:(1-2)。
10.进一步地,步骤s2中所述nacl溶液为水溶液,浓度为1mol/l。
11.进一步地,步骤s2体系中各成分的最终浓度为:ctab:0.01-0.1mol/l,nacl:0.01-0.4mol/l,镧盐:0.1-0.5mol/l。
12.进一步地,步骤s3中所述nh4hco3溶液的浓度为0.5-1.5mol/l;所述nh4hco3的加入量是镧盐摩尔量的1.0-1.2倍。
13.进一步地,步骤s3中所述nh4hco3加入时间控制在0.5-1.5h。
14.进一步地,步骤s3中所述洗涤操作中,使用水和无水乙醇分别洗涤滤饼2-4次。步骤s3中所述干燥操作中,干燥温度为80-100℃、时间为1-5h。
15.本发明使用乙二醇、聚乙二醇800作为反应溶液,同时作为分散剂,避免反应过程中碱式碳酸镧的团聚、凝结;ctab(十六烷基三甲基溴化铵)在氧化镧合成中起双重,首先,它可以进行形貌控制作为修饰剂,可以通过控制其浓度阻止还原过程中晶体某些面的生长,从而促进其他面的生长;另一方面它可以作为稳定剂使碱式碳酸镧粒子分散开,阻止碱式碳酸镧的团聚。
16.由于采用以上技术方案,本发明的有益效果包括:本发明提出了一种简单、低成本的乙二醇、聚乙二醇800混合多元醇法制备超细氧化镧的制备方法;本发明以ctab为表面活性剂,用nacl控制反应速率进而控制氧化镧尺寸,用nh4hco3和氨水作为沉淀剂制备了尺寸可控的超细氧化镧,制备的氧化镧为球形,氧化镧尺寸可根据需要控制在0.01-0.18μm范围内,粒度均匀度较高。
具体实施方式
17.下面结合实施例作进一步说明,但本发明不局限于这些实施例。
实施例1
18.s1:将300g氧化镧加入到1.5mol/l的硝酸溶液中制备1.0mol/l的硝酸镧溶液。
19.s2:将硝酸镧溶液加入到乙二醇、聚乙二醇800的混合溶液(乙二醇和聚乙二醇800的混合体积比为1:1)中,搅拌混合后加入ctab粉末固体和浓度为1mol/l的nacl水溶液;体系中各成分的最终浓度为:ctab:0.05mol/l,nacl:0.1mol/l,硝酸镧:0.25mol/l。
20.s3:将步骤s2的混合液加热至70℃,并缓慢加入1.0mol/l的nh4hco3溶液,nh4hco3加入时间控制在1h,nh4hco3的加入量是la(no3)3摩尔量的1.1倍,搅拌混均,然后加入25wt%的氨水调节ph=6,搅拌2h,使溶液中出现白色沉淀物,过滤,用去离子水洗涤滤饼3次(每次洗涤使用150g去离子水)、用无水乙醇洗涤滤饼3次(每次洗涤使用150g无水乙醇),90℃下干燥3h得到粉末状碱式碳酸镧laohco3。
21.s4:将碱式碳酸镧在850℃的马弗炉中煅烧4h,即得超细la2o3粉末。
实施例2
22.s1:将300g氧化镧加入到2.0mol/l的硝酸溶液中制备1.5mol/l的硝酸镧溶液。
23.s2:将硝酸镧溶液加入到乙二醇、聚乙二醇800的混合溶液(乙二醇和聚乙二醇800的混合体积比为1:1.5)中,搅拌混合后加入ctab粉末固体和1mol/l的nacl水溶液;体系中各成分的最终浓度为:ctab:0.01mol/l,nacl:0.01mol/l,硝酸镧:0.5mol/l。
24.s3:将步骤s2的混合液加热至60℃,并缓慢加入0.5mol/l的nh4hco3溶液,nh4hco3加入时间控制在0.5h,nh4hco3的加入量是la(no3)3摩尔量的1.0倍,搅拌混均,然后加入25wt%的氨水调节ph=7,搅拌1h,使溶液中出现白色沉淀物,过滤,用水洗涤滤饼4次(每次洗涤使用150g去离子水)、用无水乙醇洗涤滤饼2次(每次洗涤使用150g无水乙醇),80℃下干燥1h得到粉末状碱式碳酸镧laohco3。
25.s4:将碱式碳酸镧在800℃的马弗炉中煅烧2h,即得超细la2o3粉末。
实施例3
26.s1:将300g氧化镧加入到1.0mol/l的硝酸溶液中制备0.5mol/l的硝酸镧溶液。
27.s2:将硝酸镧溶液加入到乙二醇、聚乙二醇800的混合溶液(乙二醇和聚乙二醇800的混合体积比为1:2)中,搅拌混合后加入ctab粉末固体和1mol/l的nacl水溶液;体系中各成分的最终浓度为:ctab:0.1mol/l,nacl:0.4mol/l,硝酸镧:0.1mol/l。
28.s3:将步骤s2的混合液加热至80℃,并缓慢加入1.5mol/l的nh4hco3溶液,nh4hco3加入时间控制在1.5h,nh4hco3的加入量是la(no3)3摩尔量的1.2倍,搅拌混均,然后加入25wt%的氨水调节ph=5,搅拌3h,使溶液中出现白色沉淀物,过滤,用水洗涤滤饼2次(每次洗涤使用150g去离子水)、用无水乙醇洗涤滤饼4次(每次洗涤使用150g无水乙醇),100℃下干燥5h得到粉末状碱式碳酸镧laohco3。
29.s4:将碱式碳酸镧在900℃的马弗炉中煅烧6h,即得超细la2o3粉末。
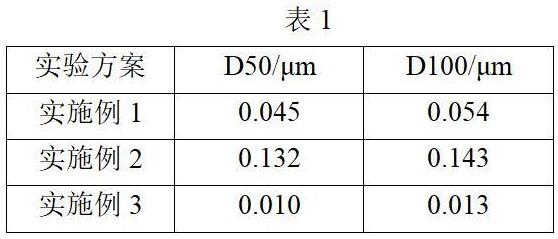
30.表1为实施例1-3制备的超细la2o3粉末的粒度数据表。
31.实施例4
32.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.01mol/l。
实施例5
33.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.05mol/l。
实施例6
34.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.2mol/l。
实施例7
35.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.3mol/l。
实施例8
36.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.4mol/l。
37.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中不添加nacl水溶液。
38.氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中加入nacl水溶液后,体系中nacl的最终浓度为0.5mol/l。
39.表2为步骤s2体系中不同nacl最终浓度的实施例或对比例制备的氧化镧的粒度数据表。
[0040][0041]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中将nacl替换为nabr,体系中nabr的最终浓度为0.1mol/l。
[0042]
本对比例3制备的氧化镧的粒度:d50=0.058μm,d100=0.079μm。
[0043]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中将nacl替换为nabr,体系中nabr的最终浓度为0.4mol/l。
[0044]
本对比例4制备的氧化镧的粒度:d50=0.028μm,d100=0.047μm。
[0045]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中将nacl替换为nabr,体系中nabr的最终浓度为0.5mol/l。
[0046]
本对比例5制备的氧化镧的粒度:d50=0.027μm,d100=0.045μm。
[0047]
(1)将氧化镧加入到1.6mol/l硝酸溶液中制备1.5mol/l的la(no3)3溶液;(2)将la(no3)3溶液加入到乙二醇、聚乙二醇600的混合溶液中,充分搅拌至溶液溶清后加入聚乙烯吡咯烷酮(mw=130 000)和nacl溶液,充分混合均匀;其中,乙二醇和聚乙二醇600的体积比为1:1;体系中各成分的最终浓度为:聚乙烯吡咯烷酮:0.1mol/l,nacl:0.1mol/l,la(no3)3:0.5mol/l;
(3)将步骤(2)的混合液加热至75℃,并缓慢加入1.5mol/l的(nh4)2co3溶液,(nh4)2co3的加入用时控制在1.5h,(nh4)2co3的加入量是la(no3)3摩尔量的1.2倍,加入(nh4)2co3溶液时搅拌速度控制在100r/min;加入(nh4)2co3溶液过程中出现白色沉淀物,反应2h后过滤、干燥得到laohco3粉末;(4)将laohco3粉末在900℃的马弗炉中焙烧5h,即得白色球形la2o3粉末。
[0048]
制备的氧化镧粉末的粒度,d50=0.049μm,d100=0.070μm。
[0049]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中将表面活性剂ctab替换为pvp(mw=13 000),体系中pvp的最终浓度为0.05mol/l。
[0050]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中将表面活性剂ctab替换为pvp(mw=58 000),体系中pvp的最终浓度为0.05mol/l。
[0051]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中不添加ctab,部分氧化镧发生了团聚现象。
[0052]
表3为实施例1或对比例7-9制备的氧化镧的粒度数据表。
[0053][0054]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中:乙二醇和聚乙二醇800的混合体积比为1:0。
[0055]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中:乙二醇和聚乙二醇800的混合体积比为1:0.5。
[0056]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中:乙二醇和聚乙二醇800的混合体积比为1:3。
[0057]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中:乙二醇和聚乙二醇800的混合体积比为0:1。
[0058]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s2中:将聚乙二醇800替换为聚乙二醇600,乙二醇和聚乙二醇600的混合体积比为1:1。
[0059]
表4为实施例1和对比例10-14制备的氧化镧的粒度数据表。
[0060][0061]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s3中:直接一次性加入nh4hco3,加入的nh4hco3的加入量是la(no3)3摩尔量的1.1倍。最终制备的氧化镧发生了明显的团聚现象。
[0062]
氧化镧的制备方法与实施例1相同,其不同之处在于,步骤s3中:nh4hco3加入时间控制在0.5h。
[0063]
氧化镧的制备方法与实施例1相同,其不同之处在于步骤s3不添加氨水,具体步骤s3的操作如下:s3:将步骤s2的混合液加热至70℃,并缓慢加入1.0mol/l的nh4hco3溶液,nh4hco3加入时间控制在1h,nh4hco3的加入量是la(no3)3摩尔量的1.1倍,搅拌混均,然后再加入0.2倍la(no3)3摩尔量的nh4hco3;搅拌2h,使溶液中出现白色沉淀物,过滤,用去离子水洗涤滤饼3次(每次洗涤使用150g去离子水)、用无水乙醇洗涤滤饼3次(每次洗涤使用150g无水乙醇),90℃下干燥3h得到粉末状碱式碳酸镧laohco3。
[0064]
表5为实施例1和对比例15-17制备的氧化镧的粒度数据表。
[0065][0066]
以上所述,仅为本发明的优选实施例,并不用于限定本发明;但对于本领域的普通
技术人员在不脱离本发明技术方案范围内,可利用以上所揭示的技术内容而作出的些许更动、修饰与演变的等同变化,均为本发明的等效实施例;同时,凡依据本发明的实质技术对以上实施例所作的任何等同变化的更动、修饰与演变等,均仍属于本发明的技术方案的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1